机器学习模拟铝原子运动,助力计算材料科学自
编译/文龙
最近,美国洛斯阿拉莫斯国家实验室开发了一种用于模拟铝等材料中原子的运动的机器学习(ML)方法。这种自动的“原子间电势发现”方法可以改变计算材料发现的领域。
该研究以Automated discovery of a robust interatomic potential for aluminum为题发表在本周的《自然通讯》(Nature Communications)杂志上。

如果拥有足够的训练数据,机器学习模型就能给开发提供具有可移植性的模型的多种可能性。这在材料物理、化学和生物学中都有许多应用,像是晶体结构预测、药物开发、材料老化和高应变速率变形的模拟都将受益于更好的原子间电势。因此,用机器学习(ML)发现原子间电势这一课题正在迅速发展。
要想最大程度地提高这些机器学习模型的总体准确性,必须设计一个高度多样化的数据集来训练模型,但问题是ML模型最先需要什么样的训练数据并不是先验的。该团队提出了一种自动的「主动学习」(active learning, AL)方法,用于迭代地构建训练数据集,并通过建立潜在的铝元素(ANI-Al)进行展示。论文的主要作者贾斯汀·史密斯(Justin Smith)表示:「这样的机器学习模型旨在以较小的计算成本来模拟高精度量子模拟的结果。」
AL的原始输入是一个初始的训练数据集,该数据集由随机(无序)原子构型的傅立叶变换组成,分子动力轨迹也被初始化为随机的密度。在每次迭代中,该方法都使用当前最佳的机器学习模型进行原子模拟。当遇到模型不了解的新物理情况时,将通过量子模拟收集新的参考数据,并对ML模型进行重新训练。通过这个过程,主动学习程序会收集许多不同类型的原子构型数据,包括晶体结构以及晶体中的缺陷。
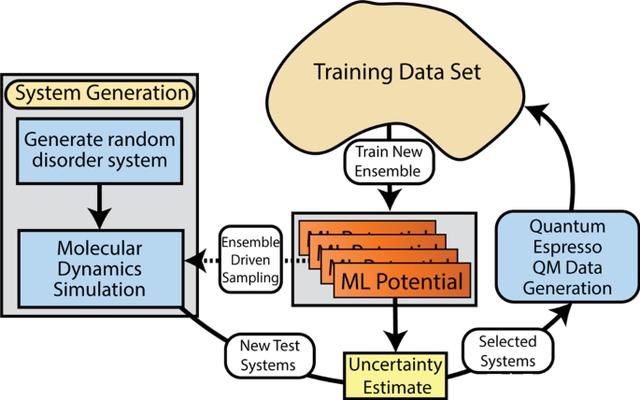
图示:同时运行的多个主动学习周期,使用同步点将新数据合并到单个全局数据集中。(来源:论文)
ANI模型是用于建模原子间电势的神经网络结构,以原子配置(核位置以及种类)作为输入,每个原子都会单独评估,再进行加和得到总能量。该团队用铝元素创建了铝在结晶相和熔融相中的ANI模型,命名为ANI-Al。为了提高预测质量,他们采用了八个神经网络的集合平均作为总能量,其中每个神经网络使用相同的数据进行训练,但模型的参数互相独立且随机初始化。
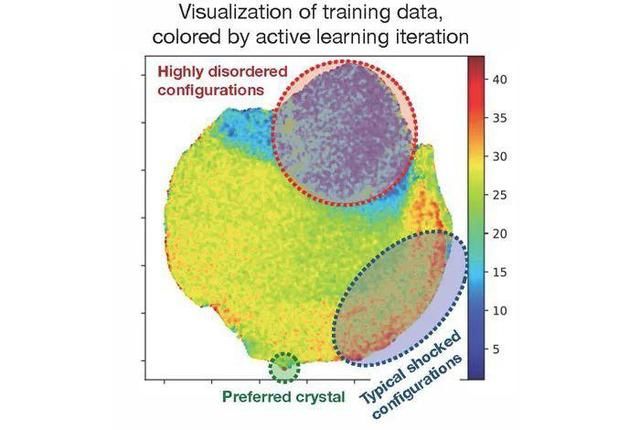
图示:自动化的数据集生成可提供高度多样化的原子位置采样,以训练准确而通用的机器学习模型。(来源:论文)
实验从多个维度进行比较,结果显示机器学习模型实现了对每个原子仅有几meV的电势误差,这是前所未有的精度。
洛斯阿拉莫斯国家实验室的项目负责人Smith说:「这种方法有望成为从基本原理研究材料损坏和老化的重要基础。模拟相互作用的原子动力学是理解和开发新材料的基石。机器学习方法为计算科学家们提供了新的工具,可以准确有效地进行原子模拟。」
论文链接:https://www.nature.com/articles/s41467-021-21376-0#Abs1
项目地址:
文章来源:《冶金自动化》 网址: http://www.yjzdhzz.cn/zonghexinwen/2021/0302/374.html